蛋白质生物技术药物的稳定性对活性药物开发的成功或失败至关重要。 蛋白质稳定性对生产、制造、制剂、长期储存、输送给患者和疗效非常重要。 高度稳定的蛋白质在制造过程中可能会遇到的问题更少,根据成本效益,更易于在制剂开发和储存期间保持功能性,而不会发生化学改变或聚集。 在生物技术药物开发的“质量源于设计”(QbD) 方法中,稳定性表征是所有候选药物的“开发可行性”和“成药性”评估以及流程开发和生产的一部分。 稳定性数据还整合到用于生产支持、生物技术药物可比性和生物相似性评估的高级结构 (HOS) 表征和“指纹图谱”中。 此外,蛋白质 HOS 表征正在成为新生物技术药物和生物相似性注册申报中的预期标准。
由于蛋白质的复杂性,生物物理工具对于生物技术药物产品的完全表征非常重要。 现在,有多种生物物理工具用于蛋白质稳定性的评价,包括但不限于:圆二色光谱 (CD)、动态和静态光散射法 (DLS 和 SLS)、尺寸排阻色谱-多角度光散射 (SEC-MALS)、傅里叶变换红外光谱 (FTIR)、分析性超滤 (AUC)、尺寸排阻色谱法 (SEC)、差示扫描荧光 (DSF)、内源荧光 (IF) 和差示扫描量热法 (DSC)。
虽然所有这些生物物理试验在生物技术药物开发中都具有非常重要的作用,但通过 DSC 表征热稳定性是最关键的。 在一篇 2015 年有关用于单克隆抗体高级结构表征的生物物理技术文章中,Gokarn 等 表明: “DSC 仍是在给定缓冲液条件下评价蛋白质热力学稳定性的无与伦比的技术”[1]。
本白皮书旨在解释如何使用 DSC 在候选药物选择过程中对蛋白质生物技术药物(主要是抗体)进行表征,以继续进入开发阶段。
DSC 是用于表征蛋白质、核酸、脂类和其他生物聚合物的热稳定性和构象稳定性的微量热法 [2-7]。 DSC 测量热容作为温度函数。 DSC 仪器用于蛋白质表征,在本白皮书中描述为“功率补偿”仪器,其中溶液中的生物聚合物置于固定样本池中,而对应缓冲液置于匹配的参比池内。 来自样本池的热容 (Cp) 信号与来自参比池的信号进行比较。 随着两个池中的温度升高,参比池和样本池之间的温度差将会持续测量并校准为功率单位。 DSC 是一种“强制降解试验,随着蛋白质暴露到不断升高的温度中,蛋白质开始去折叠,且蛋白质的 Cp 升高(图 1)。
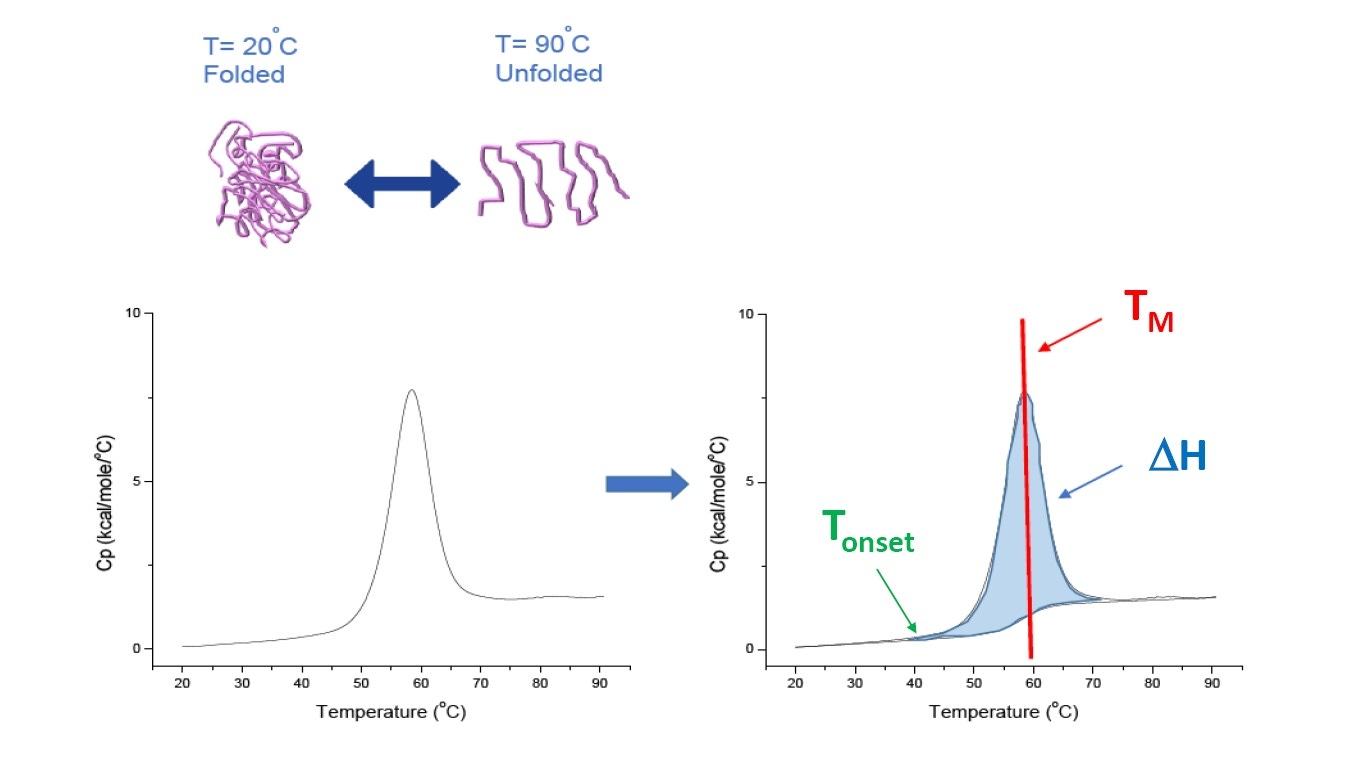
图 1:DSC 的工作原理。热容 (Cp) 随蛋白质热变性变化。 DSC 实验开始时的温度下,蛋白质主要处于其自然构象中折叠的状态。 随着温度升高,在某些点蛋白质将开始去折叠/变性 (Tonset)(热分解起始温度)且 Cp 升高。 在 50% 的蛋白质处于自然构象而 50% 已变性的温度下,Cp 将达到其最大值 - 这就是热转变中点或 TM。 高于 TM,蛋白质将主要发生变性,且在 DSC 实验结束时,所有蛋白质都将处于去折叠构象。 用于 DSC 的实验参数包括 Tonset、TM 和去折叠焓 (ΔH)。
DSC 直接测量热容改变、不需要任何荧光或任何其他标签或探针。 热转变中点 (TM),也称为熔解或变性温度,其中 50% 处于自然(折叠)构象,而 50% 处于变性构象。 TM 被看作 DSC 热谱图的“峰”(图 1)。
TM 被认为是热稳定性的良好表示 – TM 越高,蛋白质的热稳定性越好。 多结构域蛋白质(如抗体)通常在 DSC 热谱图上有多个峰,因此可确定多个 TM (示例见图 2)。
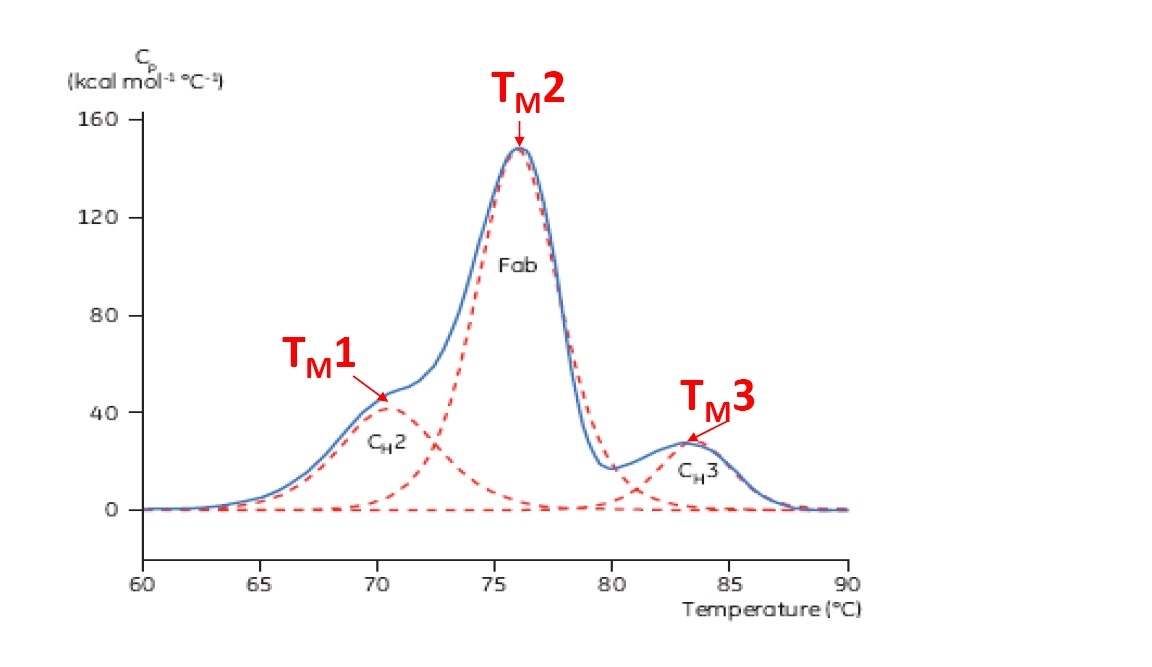
图 2:单克隆抗体的代表性 DSC 热谱图,已确认 CH2、Fab 和 CH3 结构域。 红色虚线为每个结构域转变的去卷积峰,标名三个 TM。
DSC 提供其他有用参数,这些参数可用于表征和排序蛋白质稳定性,包括由曲线下面积测量得出的去折叠焓 (ΔH)。 蛋白质去折叠是一个吸热过程,这是由于保持蛋白质折叠的二级非共价键断裂需要吸收热量。 DSC 还确定 Tonset(去折叠起始)、∆Cp(去折叠的热容改变)、T1/2(1/2 峰高度处的宽度,指示去折叠热谱图的形状)。 DSC 分析可包括确定这些参数的任何组合。
马尔文仪器提供 MicroCal VP-Capillary DSC 系统 [8,9],设计用于 TM 筛选和热力学表征溶液中蛋白质和生物聚合物的自动化 DSC。
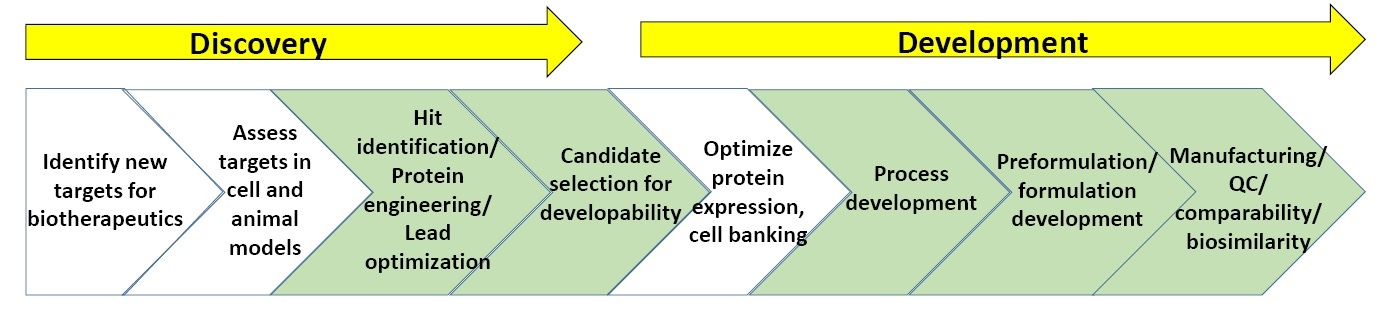
图 3:生物技术药物发现和开发流程的总体方案。
图 3 显示生物技术药物发现和开发中涉及的表征流程的总体方案。 以下绿色部分显示包括稳定性实验的生物物理表征的最常用领域。 在本白皮书结束部分为有关生物技术药物发现和开发的“补充阅读资料”列表。
氨基酸序列、或一级 (1°) 结构是多肽链和蛋白质结构的最基本组件。 了解和表征蛋白质的三维结构(也称为高级结构 [HOS])非常重要。 蛋白质 HOS 有三个水平: 二级 (2°),是指蛋白质初级结构的局部折叠方式,包括 α-螺旋、β-折叠、β-转角和无规则卷曲; 三级 (3°) 是指由大量二级结构元素构成的蛋白质三维结构;而 四级 (4°),描述由两条或更多相同或不同的多肽链相互作用构成的结构。
为创造所需生物技术药物,科学家在候选药物选择过程中最初寻找已显示出高稳定性的生物分子,而随后他们可能需要通过蛋白质工程来推断提高的稳定性。 在纯化过程中,通常要从蛋白质稳定、正确折叠和有活性的条件中去除蛋白质,因此使用正确的缓冲液、助剂、纯化方法和储存条件,以尽可能在此过程中保持蛋白质稳定,这至关重要。
如热、化学品、pH 改变、压力、混合和高浓度等的应激源在生物技术药物生产和制剂过程中频繁发生,当蛋白质分子暴露在这些应激源下时,其构象会偏向形成变性(去折叠)蛋白质。 配制的皮下注射 (SC) 蛋白质药物在非常高的蛋白质浓度时(超过 100 mg/mL)在其密封容器(如小瓶或载药注射器)内必须稳定并不受影响,时间通常为几年。 溶液中的蛋白质还对如脱酰胺作用和氧化作用的修饰敏感,这类作用可导致变性、无活性蛋白质。
就蛋白质生物技术药物而言,变性和其他修饰可导致聚集形成,进而可能造成药品疗效降低/功能性降低。 更重要的是,蛋白质聚集在可能引起患者致命的免疫反应中发挥作用。 使用稳定蛋白质药物可使生产更具成本效益,并带来更加成功、安全和有效的药物产品。
DSC 可以测量分子构象稳定性和三级、四级结构的改变(蛋白质热变性时发生),以及有关内在和外在因素如何影响蛋白质稳定性。 DSC 被认为是蛋白质生物技术药物表征中的最佳、最全面的定量热稳定性分析,并常用作长期稳定性的预测指标[1,10-14]。 来自 DSC 的 TM 是经常在候选药物选择中用于稳定性排序(开发可行性)、制剂筛选和流程开发的参数。 蛋白质越稳定,其 TM值越高。 来自 DSC 的焓 (∆H)、Tonset、T1/2和 ∆Cp 也用于稳定性排序、DSC 数验证据、蛋白质去折叠定量分析以及高级结构“指纹图谱”[10-14]。
如果分析的蛋白质相同或高度相似,定义溶液条件下蛋白质的 DSC 分析可重复并定量(图 4)。 即 DSC 热谱图将具有相似的轮廓,且参数(包括 TM、∆H 和 Tonset)将会位于可接受范围内[12-14]。 如果热谱图不同且 DSC 符合参数改变,这就表明存在蛋白质错误折叠、降解、聚集、溶剂差异、翻译后修饰改变或其他高级结构差异,这些可导致构象稳定性改变。
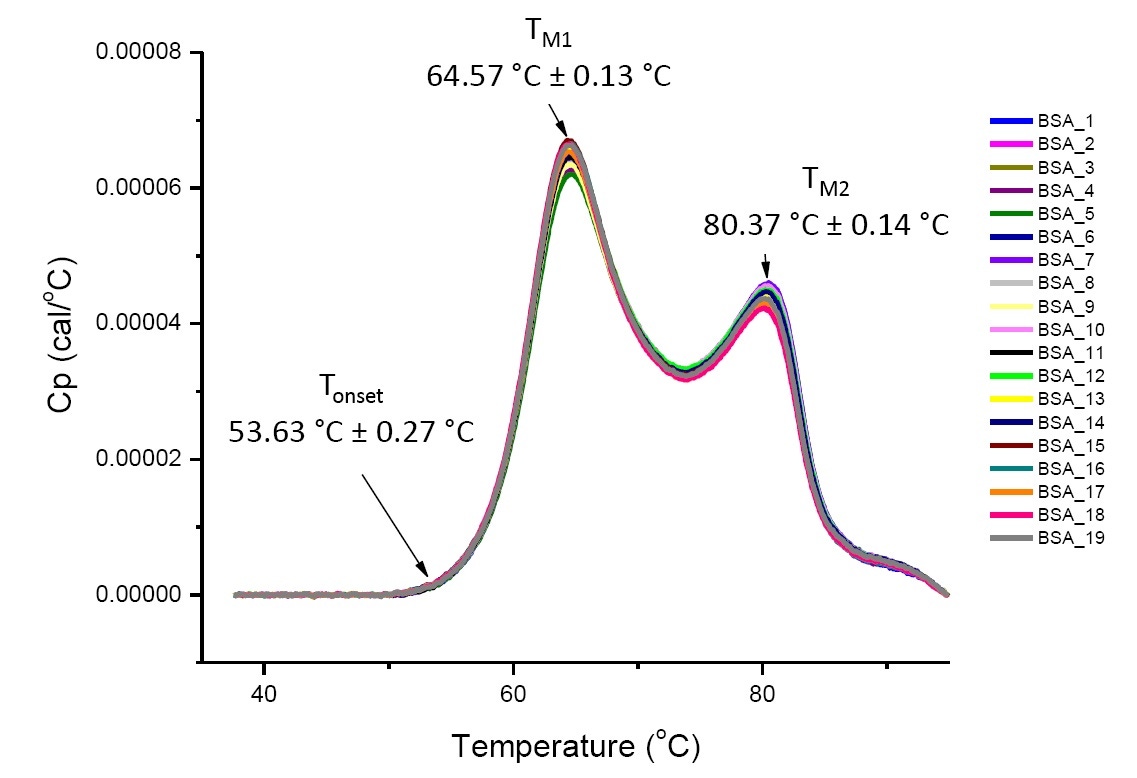
图 4. 19 个 PBS 中牛血清白蛋白的 DSC 热谱图(Sigma A1933,色谱纯化)。 DSC 数据在扫描速率标准化、缓冲液-缓冲液减法以及集成基线减法后显示。 显示 Tonset、TM1 和 TM2 的均值和标准差。
可重复和定量数据使得 DSC 成为用于在生产(包括批次间和工厂间可比性)、蛋白质变异体与修饰产物(包括由醣基化作用和氧化作用造成的结构变化)比较、以及生物相似性评价过程中评价产品的 HOS 分析中的有用工具。 DSC 数据还可在监管支持文档作为新药物和生物相似性申报的 HOS 表征中使用。 在一项生物制药行业科学家的调查中,对于在候选药物选择、制剂开发、产品表征、可比性和生物相似性中,DSC 排列为“非常有用”到“及其有用”的 HOS 生物物理工具[15]。
对于多结构域蛋白质(如抗体),DSC 热谱图存在多个去折叠转变(图 2)。 DSC 可表征和定量不同结构域,并为两个或多个转变确定 TM。 从热谱图峰获取 TM 值,并可从 DSC 数据简单确定,无需复杂数据分析。 可确定 TM 的其他生物物理试验(如 CD、IF 和 DSF) 仅可检测第一个 TM(产生于最低温度时),或多结构域蛋白质最“主要”TM。 从光谱和荧光数据提取多个 TM 需要复杂的数据拟合,且可能不具备可重现性。
与其他 TM 筛选试验比较,DSC 确实每次扫描需要更多蛋白质样本,且通量更低。 如果样本有限,一个选择是使用 DSF 或 IF 执行 TM 排序,随后选择几个样本以使用 DSC 验证 TM。 使用 DSC 验证 TM 结果非常重要,且在稳定性试验中不仅仅依赖于荧光或光谱检测 TM。 基于荧光的试验可存在干扰输出的伪影,且 TM 结果很可能会由于这些伪影移至更高(或更低)的值。 一些蛋白质和缓冲液条件与荧光不兼容,且这些方法可能检测不到 TM 差异。 最后,荧光和光谱无法确定量热焓和其他由 DSC 提供的热力学参数。
DSC 被公认为是生物制药行业中热稳定性试验的金标准,原因是 DSC:
在生物技术药物发现和开发中如何使用 DSC 数据的示例可以在本白皮书、其他马尔文白皮书和应用注释以及引用文献中查找。
在蛋白质生物技术药物的确定研究阶段,初始重点是发现与潜在药物相互作用的靶标。 一旦潜在靶标确认,工作即从找到与该靶标相互作用的药物开始,该药物将得到可产生有益临床 结果(减轻或消除疾病状态)的所需生物效应。 在确定一个或多个候选药物之后,评价这些潜在候选生物技术药物的“开发可行性”非常重要。
蛋白质药物的开发可行性是蛋白质生物物理特性及相应评价,以预测此类蛋白能否经受在细胞培养生产、纯化、制剂、包装、运输和长期储存(稳定性)过程中可能遇到的压力。 了解以下内容非常重要:
由于一旦先导候选药物选定并开始增加产量,任何改变药物或流程的尝试将导致成本上升以及增加项目停止的可能性,因此,在临床前开发开始之前,在“发现”结束阶段执行候选蛋白质药物的开发可行性分析非常重要。 在此早期做出良好选择可减少失败,并显著减少药物公司的成本以及降低具有高成本效益药物生产的成本。
在发现晚期/开发早期中的此时间点,用于生物物理试验的可用纯化蛋白量通常有限,用于开发可行性试验评价的时间也有限。 生物物理方法,如荧光、光散射法、尺寸排阻色谱和 DSC 都在此时经常使用。 其他试验,如分析性超滤和氢氘交换质谱 (HDX-MS) 也可提供有用信息。
比较候选药物来帮助寻找具有最佳稳定性的蛋白质、预测长期稳定性以及避免热稳定结构较差的分子中,DSC 进行简单早期蛋白质药物分析以排序 TM 值非常有用。 由于其在温度的作用下不稳定性更大,TM 值最低的蛋白质将考虑为预期最差的候选药物。
Doyle 等发表了一篇在发现过程中(包括候选药物选择)使用生物物理方法表征双特异性 Adnectin 候选药物的案例研究[16]。 生物物理试验包括:SEC、AUC、SEC-MALS、X 射线结晶学、SPR、DSF 和 DSC。 通过 DSC 对两个 Adnectin 候选药物的比较显示,与候选药物 A 相比,候选药物 B 的 TM 和 Tonset 都更高(表明具有更高的热稳定性)。 通过 DSC,候选药物 B 还显示出折叠可逆性;即蛋白质样本在 DSC 中冷却和再加热,而第 2 个 加热循环显示相同的 DSC 谱图。 该可逆性还表明表达水平升高以及聚集构象的趋势降低。
上市/临床试验中最大一类蛋白质生物技术药物为单克隆抗体。 单克隆抗体通常在 DSC 中显示复杂的多结构域热谱图(有关示例请参见图 2)。 最大、最主要的结构域峰为抗体的 Fab (抗原结合片段)。 CH2 和/或 CH3 构域也很常见。 不同结构域的相对位置和 TM取决于特定单克隆抗体,并可根据亚类和改造有所不同,如以下示例所示。
在 2007 年,Garber 和 Demarest 发表了一篇描述使用 DSC 表征 17 种全长治疗性抗体的文章[17,18]。 使用 MicroCal VP-Capillary DSC,这些抗体的 Fab 显示出热去折叠转变,中点 (TM) 变化范围从 57°C 至 82°C。 作者表明 IgG1 是最稳定的人亚类蛋白,随后为 IgG2 和 IgG4,且对于使用的示例,其显示出的相似亚类抗体间的稳定性的变化产生自可变结构域。 每个 V 基因的独特特征造成了 Fab 的稳定性具有较大不同。
一些抗体 Fab 可能非常稳定,而其他可能相对不稳定。 在参考文献 17 中包括的多个重组体可观察到,与 Fab TM 升高的抗体相比,来自 TM 的非常低的 Fab 稳定性与抗体表达较差/减少和聚集增多的问题相关。 结果,纯化 TM 较低的蛋白质更加困难,导致在流程开发过程中产量降低,以及储存过程中聚集的可能性增加。来自该研究的数据表明 DSC 稳定性数据在生物技术药物的候选药物选择中是有用工具[17]。
Ionescu 等在中性 pH 条件下,生成了三种(使用 MicroCal VP-Capillary DSC)人源化 IgG1 单克隆抗体及其 Fab 和 Fc 段(酶消化后)的 DSC 曲线[19]。 有一些例外,热谱图显示实验焓更大的转变,包括 Fab 片段的作用(与参考文献 17 中的结果类似)。 同样还发现表观 TM 差别显著,即使对于源自相同人种系的 Fab 片段也是如此。 Ionescu 等 提出使用去折叠焓作为识别完整 IgG1 抗体熔解曲线图中去折叠事件的重要参数。 一些 DSC 热谱图显示两个转变:第一个转变代表 Fab 片段和 CH2 结构域的去折叠,第二个转变代表 CH3 结构域去折叠。 在其他抗体中,第一个 DSC 转变代表 CH2 结构域去折叠,第二个转变代表 Fab 片段和 CH3 结构域。 还存在其他情况,DSC 曲线代表三个转变,与 CH2 和 CH3 结构域的熔解相比,其中 Fab 去折叠发生于不同的温度。 稳定性低或 Fab 片段异质性可能对长期储存或生产一致性带来问题。 作者提出完全了解 DSC 曲线的特征对于在治疗性单克隆抗体开发早期阶段的候选药物选择非常重要。
以上两篇论文描述的工作以及其他同时期发表的论文形成了后续案例研究以及使用 DSC 和其他热稳定性表征研究作为候选生物技术药物选择的基础工作的一部分。
为进一步描绘候选药物的热稳定性,在多个 pH 条件下对蛋白质加以分析来查看是否出现任何差异。 这些不同的 pH 可代表在制剂中遇到的条件和/或在药物制造过程中使用的处理缓冲液。
Jiang 等发表文章,使用选定的 HOS 表征评价两种潜在单克隆抗体药物的开发可行性(制造以及维持整体产品质量的能力)[20]。 抗体 X 和 Y 为与相同靶标作用的 IgG2 mAb,显示了类似的生物学活性。 使用生物物理技术组合(包括 DSC、近紫外圆二色谱、FTIR、DLS、荧光光谱和 SEC):他们观察:
选择酸性条件 (pH 3) 来复制在抗体纯化和病毒灭火过程中使用的缓冲液,pH 5 缓冲液模拟储存条件(制剂),而 pH 7 缓冲液在整个纯化过程中使用。
pH 7 缓冲液 (PBS) 中对两种 mAb 的 MicroCal VP-Capillary DSC 扫描显示两个转变:CH2/Fab 结构域代表第 1转变,而 CH3 结构域去折叠代表第 2转变[20]。 基于来自 DSC 的更高的 TM 以及更高的起始温度 (Tonset),mAb Y 在 pH 7 条件下热稳定性更好。 在 pH 3 缓冲液 (C3N) 中,只有一个 DSC 热转变,表明样本在起始温度时由于低 pH 已部分去折叠。 两个候选药物在 pH 3 条件下的热转变温度和焓比在 PBS 中的低很多,与预期相似。 但是,mAb X(TM 为 63.2°C)在 C3N 中显示出比 mAb Y (TM 为 48.2°C)更好的热稳定性,尽管 mAb Y 在中性 pH 条件下热稳定更好。 尽管结果非典型,但与近紫外圆二色谱数据(显示 mAb X 在 pH 3 条件下保留更多三级结构)一致。 由 pH 3 处理诱导的两组候选药物热稳定性的改变在随后 PBS 透析后完全可逆。
总体而言,DSC 结果表明两种蛋白在中性 pH 条件下都非常稳定,且 mAb Y 的热稳定性在 PBS 中比 mAb X 略高。但在 C3N 中,mAb Y 比 mAbX 低[20]。 CD 和 DSC 数据显示,在 pH 3 条件下 mAb Y 发生的结构改变比 mAb X 多。 pH 3 诱导的热稳定性改变对两种 mAb 都完全可逆,尽管 mAb Y 三级结构改变最不易恢复 与 mAb X 相比,mAb Y 在中性 pH 条件下显示出更好的热稳定性,且在 pH 5 制剂缓冲液中在 37°C 条件下储存稳定性更高。 总之,结果表明处理稳定性(对抗 pH 改变的稳固性)和储存稳定性(升高温度的影响)具有不同的驱动力。
由于 mAb Y 在 pH 7 的 PBS 中具有更高的热稳定性,在 37°C 储存过程中,在 pH 5 条件下有更高的稳定性,建议采用 mAb Y 继续研究以供进一步开发[20]。 同样,由于两个三级结构破坏的不可逆性以及低 pH 孵育后在 PBS 中观察到的自缔合,建议推进 mAb Y 开发的同时也建议开发纯化流程以尽量避免 mAb Y 暴露于 pH 3。 该案例研究表明作为候选药物选择和流程开发一部分的稳定性数据如何帮助使流程产生的不可逆聚集和构象变异最小,并最终增加过程产量[20]。
使用 DSC 热稳定性来评价和预测开发可行性的更多案例研究在 Satish 等的文章中进行了总结[21]
Tavakoli-Kenshe 等想要观察除稳定性研究以外的哪些其他指标可用于预测开发可行性[22]。 他们观察了 IgG1 和 IgG4 抗体的五种变异体,并使用旋转圆盘剪切装置(在已知固态-液态接触面产生定义的剪切条件来测量该环境中的稳定性)。 抗体基于剪切装置输出进行了稳定性排序,并与来自 MicroCal VP-Capillary DSC 的 CH2 结构域 (TM1) 的加速热稳定数据和熔解温度进行了比较。 结果表明这些技术为正交关系,基于分子间相互作用的热学方法和基于局部去折叠剪切装置稳定性揭示导致聚集的欠稳定区域。 分子模型显示对抗体结构的修饰的作用,并指示 Fc 构象和 Fab-Fc 对接在确定悬浮蛋白质稳定性中可能发挥的作用。 数据介绍了剪切测试作为蛋白质正交稳定性指标,与传统热力学方法(如 DSC)互补,允许为纯化流程选择稳定性较高的蛋白质候选药物。
在生物技术药物发现中,经常会对“靶点”分子或“亲代”蛋白质药物分子进行修饰或改造以提高其生物物理特性并使蛋白质更具开发可行性。 改造后,在候选药物选择之前,评价不同改造蛋白质的稳定性来观察改造是否导致了任何有害作用非常重要。 其中一个此类示例在马尔文应用注释中列出,其中 MicroCal VP-Capillary DSC 用作亲代抗体和两个改造蛋白质的筛选工具,以帮助预测改造后重组体的热稳定性和开发可行性[23]。 加速储存后的聚集构象(通过 SEC)也进行了研究。 TM 降低最多的改造抗体与加速稳定性筛选后聚集量最高的抗体重组体相关,可以帮助指导候选药物选择。
Demarest 等 改造了不稳定 Fab 并使用 MicroCal VP-Capillary DSC 来评估蛋白质稳定性的提升,并作为展示[18, 24]。 在开发抗体和抗体片段稳定化策略的努力中,他们选择可以识别从最初人类来源分离的破伤风毒素 (αTT) 中效果较差的 Fab,并采取诱变使 Fab 再次获得稳定性。 作者随机选择 45 个残基位置,并挑选了约 4500 个单克隆并在表达介质中培养。 含有表达 Fab 的上清在三个升高的温度下(70°C、72°C 和 74°C)进行热稳定性研究。 显示出热稳定性增强的变异体再次进行耐热筛选重复以确定其特性(有关稳定化突变的完整列表请参见参考文献 24)。 库中大约 1% 的变异体表现出热稳定性增强。 14 个“靶点”位于 VH 结构域,其余 4 个位于 VL 结构域。 这个结果提示,天然 Fab 的稳定性是受限于 VH 的临界稳定性的。 令人惊讶的是,在约 2000 个恒定区库成员中,未发现任一突变能从整体上稳定 Fab。 作者推测在恒定区范围内发生了稳定突变,但 VH 结构域的有限稳定性控制 Fab 去折叠的温度,并限制观察到这类事件的能力。
在 αTT VL 结构域内发现了 4 个稳定突变[24]。 VK4 的共有残基 W50 突变成丙氨酸(VK1 的共有残基)或组氨酸是非常稳定的。 组氨酸非常少见于人类 κ 可变区内的第 50 位氨基酸,但人类 λ 可变区内却常常发现。 这个残基接近 VH/VL 结构域的交界。 作者推测,它对 Fab 稳定性的贡献可能与 VH 结构域的支持有关,因为 VH 结构域对 αTT Fab 稳定性的限制影响特别显著。 但是,有关分离 VL 结构域的研究结果则相反。
我们构建了 12 个重组体,它们分别包含初步筛选中鉴定出的 3 个到 11 个稳定化突变[24]。 合理地得出各种组合,以确定每种突变对 Fab 稳定性的表观贡献。 在平行转化/表达实验中,引入多个稳定突变到 αTT Fab 增加了 Fab 的表达,表明更加稳定的重组体可使细胞中的表达增多。 最佳的重组体与野生型相比,表达量稳定地提高了 3 倍以上。 每个 Fab 的稳定性通过 DSC 和圆二色光谱 (CD) 加以评价。 测量 TM 值用于表观 Fab 稳定性的排序。
一个重要考虑是这些突变可能对 αTT Fab 的功能性抗原结合具有潜在影响。 热稳定突变源于最初利用定量 ELISA 检测 CL 结构域以及 CH1 C 端的组氨酸标签的筛选。 作者在功能性 ELISA 中观察到,与野生型相比,热稳定突变体加强了 αTT Fab 的表观亲和力[24]。 每个 Fab 变异体的功能强弱与来自 DSC 的 TM 所描述的稳定性直接相关。 将功能活性和蛋白质表达与来自 DSC 的 TM 关联的能力表明选择最稳定的重组体可提升开发可行性。
Seeliger 等将计算设计策略整合到系统性抗体修饰中,用于在体外显示出有聚集倾向的抗体的修饰[25]。 通过生物物理方法(包括 MicroCal VP-Capillary DSC)以及长期稳定性实验进行评价,野生型 (WT) mAb1 改造获得的一系列紧密相关的抗体显示出稳定性增加。 如 Demarest 等 在上述与 αTT Fab 有关的示例中所提到,与野生型候选药物相比,突变蛋白表达水平也升高了[24]。 总之,在本研究中使用的实验数据和计算数据表明计算方法可如何用于指导抗体优化以增加稳定性。
原始野生型蛋白质的 DSC 显示68°C 的 TM 为热谱图上的第一个转变(识别为 Fab 结构域)。 在重链或轻链突变体的 DSC 热谱图中,来自 DSC 的第一个热转变 (Fab) 的 TM升至 68.9°C - 72.8°C。 在带有改造后重链和轻链的突变体中,在 70.5°C 时观察到第一个吸热,其最大峰 (Fab) 在 83.5°C 处。 在去除重链和轻链中的预测“不稳定性”后,抗体 Fab 去折叠的 TM 升高了约 16 K。 当去折叠通过色氨酸荧光检测到后,观察到了相同趋势[25]。
计算机模拟预测到大多数引入的突变增加了热力学稳定性。 这些结果与在 DSC 和直角光散射 (RALS) 测量获得的结果一致。 两个实验都表明随着突变数量增加,稳定性持续增加。 从两个实验中,作者还发现改造轻链对稳定性产生显著影响,这与每个突变体热稳定性的计算得出的改变一致[25]。
mAb1 的所有工程化变异体表达的抗体效价均高于 50 mg/L,而野生型 mAb1 抗体效价低于 1 mg/L。 这些仅存在一条改造链的变异体表达水平的显著升高,这无法从 DSC 结果预测,DSC 结果显示这些变异体的稳定性略微升高。 但是,计算表明单个免疫球蛋白结构域的稳定性通过引入突变得到了提升。 在涉及加热的实验中,每个单独链稳定性的作用有限是可信结果,表明观察到的作用是动力学而不是平衡的热力学作用。 只要一个结构域去折叠,复合物形成的稳定作用小时,导致第二条链也开始去折叠。 对于体内蛋白质生成和折叠,存在一个稳定折叠的结构域用作欠稳定链的成核点,这足以显著增加成功折叠抗体的量[25]。
从开发生物技术药物的公司角度出发,逐步提升的蛋白质稳定性(来自 DSC 和其他生物物理试验)是否转化为逐步提升的长期稳定性和提升的有效期? 作者在 40 °C 的条件下对改造变异体实施了加速稳定性研究,他们观察到长期稳定性得到显著提高(由来自 SEC 的单体含量评价)。 在持续 4 个月的整个测试中,完全改造的变异体也最稳定[23]。 可变结构域的热力学稳定性确实是长期稳定性的重要因素。 但从作者的数据以及其他发表数据来看,热力学稳定性和有效期无法在每个案例中都容易地加以关联,且可能需要进行调整以提升在制剂阶段的稳定性[25,26,27,28]。
最近发表的文章描述了合理改造以及使用 DSC 和其他生物物理工具来表征蛋白质稳定性,包括:
国家标准与技术协会单克隆抗体 (NISTmAb) 参比材料通过常用于 mAb 候选药物的 HOS 生物物理工具进行了开发可行性评价[35]。 NISTmAb 不是治疗分子,且 NISTmAb 的开发可行性评估类似指定类别、代表性 IgG1 mAb 进行[36]。 NISTmAb 在不同缓冲液条件下使用电泳、SEC-MALS、DLS 和 DSC 方法进行了稳定性和完整性评价。 PBS 缓冲液中 NISTmAb 样本 的热稳定性通过 MicroCal VP-Capillary DSC 确定。 NIST 样本显示三个主要热转变,分别为 71.2°C 处的 CH2 结构域,84.1°C 处的 CH3 结构域,以及 88.9°C 处的 Fab 结构域。
与 IgG1 分子的 DSC 曲线相比(参考文献 17),NIST Fab 更稳定 (88.9°C);因此,可以推断该分子具有更好的 HOS 特性[36]。 基于完整的生物物理分析数据包,NISTmAb 拥有良好的开发可行性特征,且压力条件下在分子中观察到的改变并不代表在开发流程的后期阶段通过正确制剂和制造无法降低风险。
本白皮书中呈现的结果明确表示纳入 DSC 作为候选生物技术药物选择过程中的生物物理稳定性实验的重要性和有效性。 使用 DSC 结果以及其他稳定性实验的结果,生物制药公司可明智选择最稳定和最具开发性的候选药物,即最可能生产和纯化,且在其最终制剂以及药品中不易出现长期稳定性和聚集问题的蛋白质。 这会转化为更具成本效益的药物生产,最终药物制剂更易保持活性、稳定,并处于正确折叠的构象。
Analytical Techniques for Biopharmaceutical Development, R. Rodriguez-Diaz, T. Wehr, S. Tuck (eds.), Taylor & Francis, New York USA (2005).
Biophysical Characterization of Proteins in Developing Biopharmaceuticals, D.J. Houde, S.A. Berkowitz (eds.), Elsevier, Amsterdam, Netherlands (2015).
Biophysical Methods for Biotherapeutics: Discovery and Development Applications, T.K. Das (ed.) John Wiley & Sons, Hoboken NJ USA (2014).
Biophysics for Therapeutic Protein Development, L.O. Nahri (ed.), Springer New York, USA (2013).
State-of-the-Art and Emerging Technologies for Therapeutic Monoclonal Antibody Characterization Volume 1. Monoclonal Antibody Therapeutics: Structure, Function, and Regulatory Space, J.E. Schiel, D.D. Davis, O.V. Borisov (eds.), ACS Symposium Series Vol 1176 (2014). doi: 10.1021/bk-2014-1176.
State-of-the-Art and Emerging Technologies for Therapeutic Monoclonal Antibody Characterization Volume 2. Biopharmaceutical Characterization: The NISTmAb Case Study, J.E. Schiel, D.D. Davis, O.V. Borisov (eds.), ACS Symposium Series Vol 1201 (2015). doi: 10.1021/bk-2015-1201.
State-of-the-Art and Emerging Technologies for Therapeutic Monoclonal Antibody Characterization Volume 3. Defining the Next Generation of Analytical and Biophysical Techniques, J.E. Schiel, D.D. Davis, O.V. Borisov (eds.), ACS Symposium Series Vol 1202 (2015) DOI: 10.1021/bk-2015-1202.