本白皮书探讨了 DSC 分析的原理,并以基本应用为例进行了说明。同时还探讨了 DSC 的优势和具体信息。
DSC 的关键优势在于其基于热测量,因此可以对天然生物分子进行表征。此外,没有光谱读数意味着样品无需在光学上透明。同时,该表征并不局限于熔解温度 (Tm),它还提供了关于生物大分子折叠所涉及的作用力以及展开机制的数据。
溶液中生物大分子的热力学不容易进行单独解释。测量值总是分子中基团之间的相互作用和相同基团与溶剂可能产生的相互作用的净效应。因此,稳定生物系统的能量是许多有利和潜在不利相互作用之差。这一事实对许多研究人员来说是一种启示,DSC 测量结果非常清楚地说明了这一点。然而,还有机会,仔细、有条不紊的方法可以开始相互作用的复杂性进行分析。还发现 DSC 具有许多其他用途,部分是因为该技术能够获得与负责稳定大分子作用力变化有关的全部热力学信息,且没有任何光学成分。DSC 可以测量与大分子质子化变化有关的简单效应如何影响热力学和稳定性。这种方法可以扩展到包括任何非共价结合的配体,从而提供多功能的结合筛选方法。在有利的情况下,这种方法可以用来确定结合常数。本文讨论了 DSC 测量的热力学背景,以及该技术在研究蛋白质单独稳定性以及蛋白质与配体相互作用稳定性中的应用。然而,这些技术同样可以转用于其他生物大分子(如核酸、脂质等)。
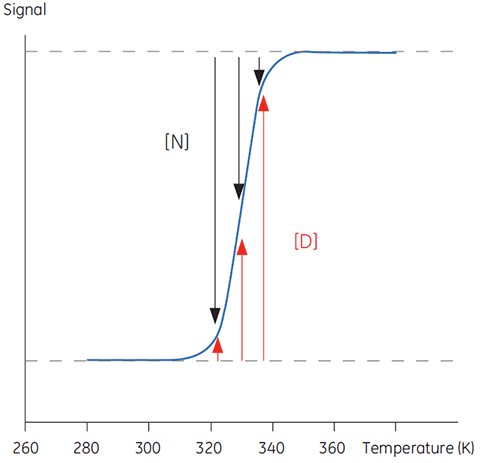
当温度上升到生理相关范围之外时,蛋白质会在结构化、天然和生物活性构象 (N) 与非结构化、变性和非活性构象 (D) 之间发生转变。一般而言,蛋白质与许多其他生物大分子(如 DNA、脂质等)以及有机聚合物都存在此类现象。如果跟踪来自报告发生此类构象转变的蛋白质的信号,就可以看到如下图所示的 S 型曲线。
|
当蛋白质结构以这种方式熔解时,分子的共价性质没有改变。只有非共价相互作用被扰乱,而且在许多情况下,当蛋白质再次被冷却时,这些相互作用将自发进行变化,产生活性天然构象。因此,N 和 D 处于可逆平衡状态,而温度是一个强度变量:

|
在图 1 中,可以观察到 N 和 D 的比例随着温度升高而向 D 的方向推动平衡改变。在任何温度下都可以定义一个平衡位置,即平衡常数 (Keq),其仅反映 N 和 D 的相对浓度。在对数坐标尺度下,该平衡常数用吉布斯自由能 (ΔG) 表示:

|

|
其中 R 为气体常数,T 为温度(开氏温度)。D 和 N 浓度相等时的温度被定义为转变或熔解温度 Tm 的中点。在此温度,Keq 等于 1,ΔG 为 0。Tm 是任何蛋白质的一个重要参数,因为代表其热稳定性。在此温度以下,天然蛋白质浓度高于变性蛋白质浓度,而在 Tm 以上,更多的蛋白质处于变性状态。
蛋白质表现出这种熔解行为的原因是其天然结构被许多本身具有温度依赖性的相互作用所稳定。通过焓值 (ΔH) 的稳定需要涉及成键作用、结构化和内能降低的相互作用,而通过熵值 (ΔS) 的稳定则反映了无序相互作用以及系统在相同能量下组织方式数量的增多。这些项与我们所熟悉的方程中的 ΔG 有关:

|
结合方程 3 和 4 并重新排列,可以得到范特霍夫方程 5,根据该方程可以绘制平衡常数随温度变化的曲线,从而得出热变性的焓和熵的简单线性关系 (lnKeqvs 1/T):
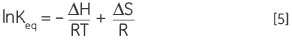
|
图 1 中的数据可以用来确定转变区温度下的 Keq。以这种方式确定的焓值被称为范特霍夫焓值,其取决于 DSC 曲线的形状。因此,它不是对反应热的直接测量。
毫不奇怪,量热法是一种直接确定蛋白质变性热的方法,该术语来源于拉丁语 calor(意思为热)和 metrium(意思为测量)。超灵敏量热仪(如 MicroCal™ VP-Capillary DSC)适用于精确测量亚毫克级材料的焓值。这些仪器使用简单、准确和可靠,使这种类型的量热测量成为任何生物物理实验室的常规组成部分。这些系统通过测量蛋白质溶液样品的热容 (Cp) 来进行工作,同时扫描温度的上升或下降。Cp 只是提高样品温度所需的能量,通常为 1 K,与基尔霍夫定律中的焓有关:

|
在扫描过程中,将测量相对于仔细匹配溶剂参比池的蛋白质过剩(差示)热容,因此这些类型的仪器被称为差示扫描量热仪。
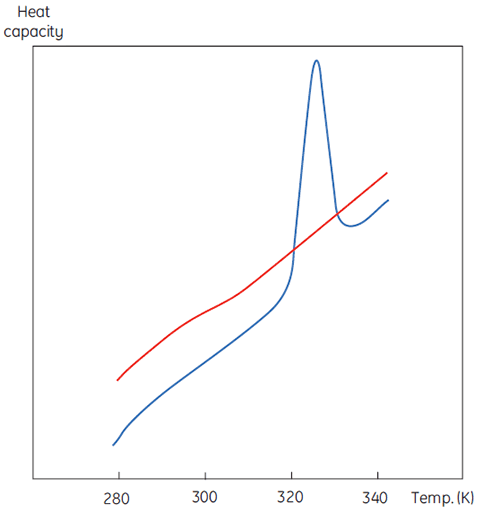
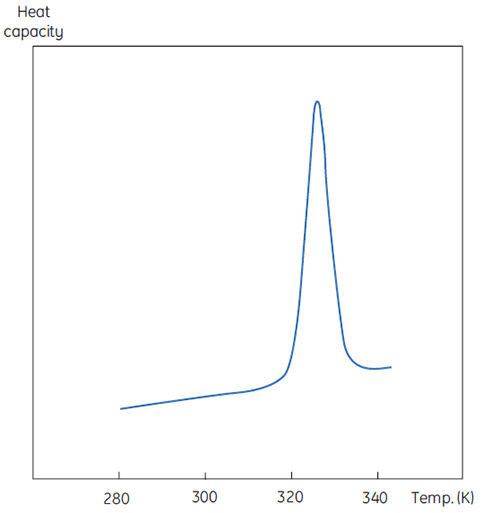
DSC 测量与图 1 所示相同,不同之处在于现在变性过程中蛋白质的性质为热容。将观察到如图 2 的构象转变。
|
|
从方程 6 中可以看出,要获得焓,必须对过剩热容函数进行积分。在积分前,必须去除在样品池和参比池都只含有溶剂时观察到的仪器基线。由于技术原因,仪器基线没有给出测量池之间的零过剩热容(红色实线,图 2 上方图)。减去基线后,将转变峰两侧的线性区域(代表蛋白质天然和变性状态的热容)外推至转变区,然后将其与转变区的进展情况合并。这些计算均由软件程序完成。最后,对所产生的峰值下面积进行积分,获得 DSC 在样品池中使蛋白质变性所需的过剩能量。只要蛋白质溶液的浓度和量热仪的操作体积已知,此能量就可以转化为每摩尔蛋白质的 ΔH(单位为卡路里或焦耳)。通常情况下,ΔHcal 用于表示直接测量的量热焓值。
MicroCal VP-Capillary DSC 是一种高灵敏度仪器,能够测量与稀溶液中蛋白质变性相关的热容的极其微小的变化。溶剂热容超过蛋白质热容很多个数量级。因此,为了进行最准确和可靠的测量,必须通过确保蛋白质样品的溶剂和参比溶液的成分完全匹配来仔细消除背景热容。透析或色谱均为合适方法,最终透析液或色谱柱流通液可以用作参比溶液。
图 2 中的数据也可以用与上面讨论的蛋白质变性时发生变化的任何其他性质相同的方式进行分析。这可以给出 Tm 和模型依赖的范特霍夫焓值;现在是 ΔHvH,用以区别于 ΔHcal。对在同一实验中获得这两个焓值进行比较,为蛋白质变性模型提供一项强有力的检测。ΔHvH 可以提供平衡中每摩尔协同单位的能量,而 ΔHcal 可以提供每摩尔蛋白质的能量。如果这些能量相同,那么蛋白质和协同单位相同,或者可以设想为具有相同的分子量,从而证实了平衡的两态假设。
在其他情形下,ΔHvH 小于 ΔHcal,表明平衡种类的平均分子量低于蛋白质的平均分子量,此时涉及中间产物 (I) 的变性方案可能更加合适:

|
在变性过程中产生中间产物的极端情况下(如蛋白质具有不同热稳定性的独立折叠域),可以观察到两种不同的转变。此时,每个转变的 ΔHvH 将反映每个域的分子量,而 ΔHcal 将基于整个蛋白质分子量反映系统变性的总能量,也就是两个转变区下的总面积。
也有可能出现 ΔHvH 大于 ΔHcal 的情况,这表明平衡种类的大小或分子量大于蛋白质。这可能是蛋白质形成了二聚体、四聚体或高阶聚集体的情况,其 ΔHvH 与 ΔHcal 之比至少在原理上可以反映结合的阶数 (n):

|
与方程 7 不同,这仍然是一个两态平衡,因为仅包含 N 和 D。ΔHvH 与 ΔHcal 之比的差异反映计算中的每摩尔浓度项基于亚单位而非低聚蛋白质。
这也提出了重要的一点,即 ΔHcal 值完全取决于样品浓度。浓度的任何误差都将直接转移到对焓值的估算中。这些因素包括浓度物理测量的误差(通常是通过吸光光谱法)、蛋白质消光系数的误差以及由低于 100% 纯材料(污染蛋白质会增加吸光度,但具有不同 Tm 值,在 DSC 中可能无法观察)或低于 100% 折叠的天然材料(变性蛋白质会增加吸光度,但已经展开)而引入的误差。因此,在解释 ΔHvH 与 ΔHcal 比值的微小差异时,必须小心谨慎。
对样品纯度和浓度的准确定量分析是 DSC 中实验设计和数据解释的最重要因素。
如果 ΔHvH 与 ΔHcal 比值表明有为低聚物变性方案(方程 8),则通过检查 DSC 测量的浓度依赖性,可以很容易地确认这一点。由于平衡涉及到浓度变化(例如单体形式),那么单体浓度将通过质量作用或勒夏特列原理影响平衡位置。因此,低聚物系统的热稳定性预期会随着浓度增加而增大,这在实践中是可以观察到 (1,2)。
在 DSC 测量中检查样品浓度的依赖性是机制的一项关键检测,如果样品可用性允许,即使计算出 ΔHvH 和 ΔHcal 比值接近 1,也值得进行该项检测。
在完成 DSC 测量后,一个简单而又可能包含丰富信息的实验是再次重新扫描同一样品。如果在重新扫描过程中观察到相同的吸热,即可得出结论,在天然和变性的平衡状态下,相互作用完全可重复。然而,在相当多的情况下,重新扫描蛋白质时没有转变或转变的幅度减少。这表明在变性机制中存在不可逆步骤:

|
在这种单分子 (U) 方案中,如脱酰胺化、脯氨酸异构化等过程可导致不可逆的修饰,并阻止再折叠。通常情况下,由于不可逆步骤存在动力学因素(速率),通过扫描至刚刚超过转变区的温度,而非 DSC 将达到的最高温度,可以提高可重复性。在较高温度下,这一速度很可能会更快。因此,进入温度越高,变性时间越长,进行的不可逆步骤越多。
不可逆步骤也可能是由变性状态的结合或聚集 (Ag) 所致:
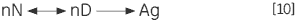
|
这将表现为初始扫描参数的浓度依赖性。在更高样品浓度下,D 的浓度会增加,因此不可逆步骤被加速,导致蛋白质的 Tm 降低。在显著和快速聚集的情况下,甚至有可能扭曲和截断变性转变与放热聚集事件,并观察到转变区域以上的非典型噪声数据,如下所示。
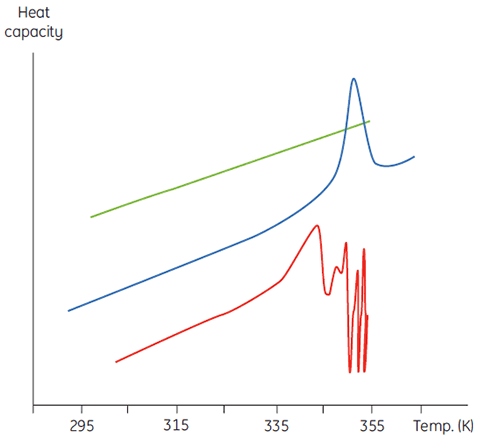
|
在这方面,量热技术不是一种宽容度很高的技术,往往是通过 DSC 发现了在类似样品浓度下进行的基于光谱的热变性实验中不明显的聚集问题。在这两种不可逆的情况下,检查 DSC 测量的样品浓度和扫描速率的依赖性很有意义。事实上,在某些情况下,此类信息可用来从初始平衡事件中获取有意义的参数,甚至可以估计不可逆速率的数值 (3,4)。
在 DSC 测量中检查扫描速率依赖性是机制的一项关键检测,如果样品可用性允许,即使系统高度可重复且平衡,也值得进行该项检测。即使在最简单的单分子平衡中(方程 1),也必须确认 N 和 D 的平衡浓度反映了该条件下系统的稳定性。这些浓度变化速度等于平衡中的正向(展开)和逆向(折叠)反应之和。因此,如果温度升高速度超过了系统变化速度,DSC 吸热的位置和形状将发生扭曲。这将导致不正确 Tm 值和异常 ΔHvH 与 ΔHcal 比值。根据经验,对于大多数蛋白质而言,60K/h 是良好的起点。
除了这种对焓的比较使用,该技术还提供了对焓本身和 Tm 值的精确测定。如前所述,Tm 反映了蛋白质的热稳定性。这不应直接等同于样品活性持久或其保存期意义上的稳定性,因为这可能存在由不可逆步骤引入的额外动力学维度。在具有完美可逆性的最简单平衡中(方程 1),理论上,蛋白质具有无限长的保存期。但很明显,在高于 Tm 的温度下,活性天然状态较少,活性水平也相应降低。在 Tm 温度以下,该体系有可能重新获得活性天然结构。在最简单的不可逆变性情况下(方程 9),当温度高于 Tm 时,非平衡步骤的速率会与从 D 到 N 的折叠速率竞争,其结果将决定蛋白质能够在多长时间内仍有机会转向活性天然状态。即使温度远低于 Tm,平衡常数意味着大部分蛋白质处于天然状态,D 到 U 的过程不可逆,表明一旦转化为 U,分子就不再处于平衡状态。为了维持适当的 [D]/[N] 比值,正如 Keq 所决定,天然蛋白质会发生变性。再一次是动力学速率决定了天然和活性蛋白质将保持多长时间。这就是说,在远低于其 Tm 的温度下储存蛋白质显然是明智的,常识告诉我们 Tm 和保存期具有相关性。在较低温度下,所有反应速度都会减慢,包括 D 到 U 的步骤。此外,如果像通常情况那样,不可逆步骤涉及两个或更多变性蛋白质的聚集(方程 10),那么 D 浓度越低,则反应速率就越慢。
将热稳定性等同于蛋白质在远离其 Tm 温度下的平衡稳定性同样是一种误导。远离 Tm 温度下推断平衡常数以及 ΔG 非常复杂,取决于下面考虑的若干参数。这种推断具有明显非线性,因此,Tm 较高的蛋白质在较低温度下的 ΔG 不一定大于 Tm 较低的其他蛋白质。
在 DSC 实验中确定的焓值适用于测量的 Tm,因为这是其确定的中点。在此温度下,ΔG 为 0,且根据方程 4,ΔS = ΔH/Tm。换言之,通过消除而非测量获得 ΔS,因此 ΔH 和 Tm 测定中的全部误差都会波及 ΔS。
如果认为这些基本的热力学量会让人对稳定蛋白质结构的作用力具有深刻的认识,那真是再好不过。毕竟,蛋白质非常像生物中的乐高™ (Lego™)。对于玩具而言,仅有几类积木块,而且其组合连接方式也只有数种。但即使存在这些限制,其仍然能够建立众多的结构和功能。在生物学中,一个蛋白质多肽仅包含有限的化学基团(积木块)可以相互作用;来自骨架羧基和酰胺基以及来自氨基酸侧链的基团。这些基团使用同样小的一组相互作用将其自身组合起来:范德华力、氢键、静电电荷吸引力和疏水作用。这些相互作用,由于其非共价性质,是处于相互作用和不相互作用两个极端之间的平衡系统,因此可以用上述热力学加以描述。所以,所确定的蛋白质变性的 ΔH 和 ΔS 必须说明这些相互作用的性质和强度。
遗憾的是,这些相互作用信息过于丰富,数量庞大。在一个普通蛋白质中,将具有数百种相互作用,这些相互作用的总和将决定整个分子的热力学特性。不可能孤立地干扰某一种相互作用。通常情况下,蛋白质在其结构中显示出高度协同性。因此,要么实现所有的相互作用而蛋白质保持天然状态,要么破坏所有的相互作用而蛋白质处于变性状态。
此外,蛋白质中的每个化学基团还可以与水和溶解的盐进行分子间相互作用以及分子内相互作用。因此,每种相互作用的热力学将反映出基团聚集状态和基团分别被水分子溶剂化状态之间的净差异,这会使情况更加复杂。
尽管不能孤立地解释 ΔH 和 ΔS 的值,但仍可以利用这些参数开展大量工作。它们均在 ΔG 为 0 的 Tm 温度下确定,所以如果我们知道其温度依赖性,则可以在所有其他温度下计算 ΔG。要做到这一点,必须知道蛋白质变性时的热容变化 (ΔCp)。由于 DSC 可以测量热容,有可能直接由这些数据确定变性时的热容变化(图 2)。事实上,由于此明显差异,在对峰面积进行积分前必须应用进展基线功能。
如图 2 所示,蛋白质的 ΔCp 较大且为正值。可以从每个 DSC 实验中获得其数值,也可以简单从蛋白质大小中确定其结果,同样准确 (5)。蛋白质越大,ΔCp 就越大,反映出其数值与蛋白质折叠到天然状态时被隐埋的非极性表面总量具有极好的相关性。
ΔH 和 ΔS 随温度的变化是基尔霍夫关系的另一种表达方式:
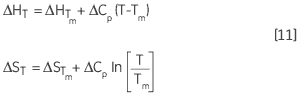
|
导致 ΔG 的温度依赖性,计算方法为:
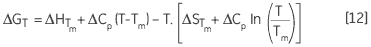
|
方程 11 还提供了另一种确定 ΔCp 的方法。如果在不同的稳定条件下测量蛋白质的 ΔH,那么 ΔH 的变化、δΔH/δT 都可以推导 ΔCp。如下文所述,只要这些实验使用适当的缓冲液,可能是对 ΔCp 最准确的估计,因为其基于对 ΔH 的一些独立测量。
通过方程 12,可以计算出任何其他关注温度下的 ΔG、ΔH 和 ΔS。这样的估计将带来一个误差,反映 Tm 和 ΔH 测量结果的原始误差以及远离 Tm 的温度外推长度。随着温度外推越长,复合误差可能变得越显著,引用时应当对其大小进行一些估计。为了比较不同系统之间或不同溶剂条件下的 ΔG、ΔH 和 ΔS,必须将测量的数据外推到共同的比较温度。
比较两种蛋白质的实验测定的变性 ΔH 毫无意义,因为这些测定结果是在其各自 Tm 值下测量的。此类比较需要利用每个蛋白质的 ΔCp 将 ΔH 数据外推到共同的温度。这个温度最好为两个 Tm 值 (T1m+ T2m)/2 之间的中点,以尽量减少外推误差。
在定性水平可以进行此类比较,并可能标记出系统中的一些粗略变化。正如前述内容,变性过程的复杂性使得对数据或其数值变化的任何详细解释都非常困难。然而,确实存在一些实验策略可能最终开始揭示特定相互作用对稳定蛋白质结构的影响。定点突变可以改变蛋白质的特定侧链,并能以非常精妙的方式使用,例如通过删除隐埋在蛋白质核心中的单个甲基。
如果亲本蛋白质和突变体的高分辨率结构信息能够补充这些突变的热力学信息,就有机会来解释这些变化。这种方法可以扩展到双或三突变体循环以确认相互作用能量的有效性,或包括溶剂的变化(H20 对比 D20)以探测氢键 (6)。
迄今为止,我们已经了解热力学数据的复杂性和信息量方面问题重重。然而,有一些方法可以利用这一点,赋予 DSC 有价值的用途。例如,蛋白质中具有质子化基团的氨基酸侧链在变性过程中可能发生 pKa 偏移。在天然状态下,它们与其他带电基团发生相互作用,而在变性的分子中,它们会与溶剂发生相互作用。pKa 只是表示该基团在其质子化和非质子化形式之间的平衡常数,如果这种平衡在变性过程中发生变化,那么质子将被释放或被蛋白质吸收。由于研究中存在缓冲液,并起到维持 pH(质子浓度)的作用,参与蛋白质变性平衡的质子将被缓冲液吸收或提供,这一过程也会产生相关的焓。这种焓值对每个缓冲液来说都是不同的,取决于其化学性质。因此,如果在相同 pH 下,在不同缓冲液中测量蛋白质变性的 ΔH,将观察到不同的数值,反映了蛋白质事件的 ΔH 和缓冲液电离的 ΔH 之和。将观察到的数值对缓冲液的电离 ΔH 作图,得到一个线性函数,其斜率(正或负)反映了与蛋白质变性有关的质子数量(所有 pKa 偏移的总和)。
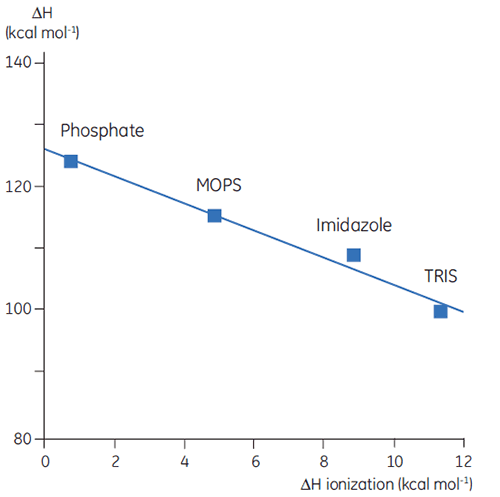
|
设计 DSC 实验时,缓冲液的选择非常重要。对于常规工作,最好使用电离焓较小或为零的缓冲液,因为这样可以消除测量中的任何电离作用。缓冲液如甲酸盐、乙酸盐和磷酸盐均为很好的选择,因为它们的电离热可以忽略不计,并且具有高纯度产品可供选择,而且价格便宜。这些缓冲液还有一个额外优点,即其 pH 随着温度变化而保持不变;显然这对 DSC 测量而言是一个重要因素。这也是它们的电离热较小的结果。
蛋白质具有可质子化的基团,其 pKa 在天然和变性状态下不同,Keq 以及 ΔG 将取决于溶液质子浓度,即 pH。这遵循质量作用或勒夏特列原理。如果变性 Keq 通过释放或吸收质子影响溶液的 pH,可以认为溶液的 pH 值会影响蛋白质的 Keq。这种影响可按照以下方程加以定量分析:
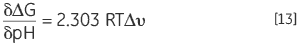
|
其中 Δυ 是质子化的变化。质子化的变化越大,每个 pH 单位的稳定性变化就越大。仅当溶液中基团的 pKa(暴露在变性状态下)和天然状态下基团的 pKa(可能与带相反电荷的基团发生相互作用而降低其 pKa,或带相同电荷的基团发生相互作用而提高其 pKa)之间存在质子化状态的潜在差异时,这种影响才会显现出来。蛋白质中典型的 pKa 偏移最多为两个单位。蛋白质通常具有钟型的稳定性 pH 曲线,准确反映了这种影响。蛋白质中酸性基团的 pKa 约为 4,碱性基团的 pKa 约为 10,因此稳定性的主要变化发生在 2 至 6 以及 8 至 12 之间。在中性 pH 下,唯一的 pKa 为组氨酸 pKa,这可能会也可能不会调节此区域的稳定性。
DSC 可用于确定蛋白质在几乎任何关注 pH 下的热稳定性。可以用 Tm 和 ΔH 来计算常见比较温度下的 ΔG,同时利用方程 13 可以分析 pH 稳定性特征,获得任何 pH 下变性的质子变化。蛋白质中带电残基的特定突变对这些特征的影响可以表明哪些基团参与了质子化行为。
把蛋白质视为简单非共价连接配体,且在天然状态和变性状态下具有不同的“亲和力”(pKa),这可能更易于理解。利用这个类比,我们可以立即想到,质量作用的效果必须适用于任何对天然和变性状态具有不同结合亲和力的配体。即使并非所有配体,绝大多数生物学相关配体都能够与其天然状态的同源蛋白质发生紧密而特异性结合,而对相应的变性状态没有亲和力。因此,当它们变性时,将配体释放到溶液中,影响浓度。质量作用影响是指当溶液中配体浓度发生变化时,将影响天然到变性的平衡,也就是蛋白质的稳定性。
该方法可以用在两个方面,首先是作为一种粗略的筛选工具,用于筛选与蛋白质结合的配体。蛋白质的热稳定性是在其自身上测量的,然后存在配体的情况下进行筛选。这些均以某种程度的过量浓度加入蛋白质,确保具有合理亲和力的配体结合可以使蛋白质上的结合位点饱和。在热稳定性增加的情况下,有证据表明在变性温度下,通过与天然状态的结合来稳定蛋白质。相反,热稳定性降低表明配体与变性而非天然状态的蛋白质结合。如果热稳定性变化较小,必须注意一些问题,因为配体也可能对离子强度或 pH 改变产生间接作用,然后可能调节稳定性。适当的控制可以应对这些可能性。
该方法第二个方面的用途是更加定量的分析,涉及测量热稳定性作为增加配体浓度增加的函数。如图 5 的数据,这些数据可用于获得配体和蛋白质之间结合常数的估计值 (7)。
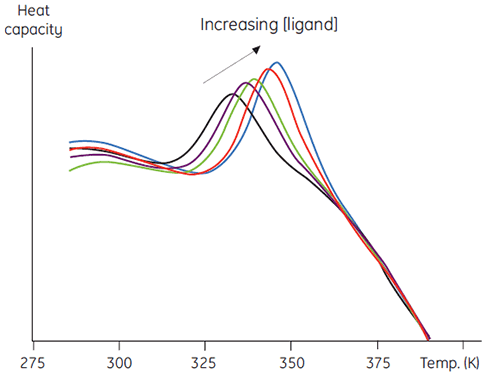
|
与其他更传统的方法相比,该方法有几个优点。该方法可用于确定常规平衡方法无法测量的非常紧密的结合常数。测量的信号为 ΔH,这是蛋白质变性的普遍特征,因此无需针对研究的每种蛋白质开发特定方法。此外,还可以使用在光谱方法中可能存在问题的高浓度配体。配体混合物可用于筛选与已知配体的竞争性结合,或用于精心设计的组合筛选方案。
最近,DSC 仪器和自动化技术的进步使我们能够生产出一种自动化的高通量 DSC,即 MicroCal VP-Capillary DSC (8)。除了使用自动化加载样品外,这些仪器的扫描速度更快,使整个测量更加迅速。在采用更高的扫描速率前,明智的做法是检查 Tm 和焓对该参数的依赖性,以确保系统处于上述的平衡状态。一旦确立,该系统可在 24 小时内采集 25 次左右的扫描,并且可以加载样品,将其置于冷藏室中,使仪器可以运行一周。
此自动化 DSC 方法是一种强大的分析工具,可用于表征蛋白质和其他生物分子的稳定性。可以快速评估影响蛋白质热稳定性的溶液条件(pH、离子强度、添加剂)。此信息可能有助于说明保存期研究或结晶试验的最佳条件。同样,与蛋白质结合从而增加其热稳定性的配体,可以在更高通量的初筛中确定其特征,或对其进行详细评估。
本白皮书的作者为 Chris M. Johnsson 博士,他目前受雇于 MRC 蛋白质工程中心(地址为 Hills Road,Cambridge,CB2 2QH,UK [英国])。电子邮件为 cmj@mrc-lmb.cam.ac.uk
Neet, K.E. and Timm, D.E. Conformational stability of dimeric proteins: Quantitative studies by equilibrium denaturation. Prot. Sci. 3, 2167-2174 (1994).
Johnson, C.R. et al. Thermodynamic analysis of the structural stability of the tetrameric oligomerization domain of p53 tumour suppressor. Biochemistry 34, 5309-5316 (1995).
Freire, E. et al. Calorimetrically determined dynamics of complex unfolding transitions in proteins. Ann. Rev. Biophys. Chem. 19, 159-188 (1990).
Lepock, J.R., et al. Influence of transition rates and scan rate on kinetic simulations of differential scanning calorimetry profiles of reversible and irreversible protein denaturation. Biochemistry 31, 12706-12712 (1992).
Myers, J.K. et al. Denaturant m values and heat capacity changes: relation to changes in accessible surface areas of protein unfolding. Prot. Sci. 10, 2138-2148 (1995)
Connelly, P.R. et al. Enthalpy of hydrogen bond formation in a protein ligand binding reaction. Proc. Natl.Acad.Sci. USA, 91, 1964-1968 (1994).
Brandts, J.F. and Lin, L.N. Study of strong to ultratight protein interactions using differential scanning calorimetry. Biochemistry 29, 6927-6940 (1990).
Plotnikov, V.V. et al. An autosampling differential scanning calorimeter instrument for studying molecular interactions. Assay Drug Dev. Technol. 1, 83-90 (2002).
Cooper, A. et al. Differential scanning microcalorimetr, in Protein-Ligand Interactions: hydrodynamics and calorimetry (Harding, S. E. and Chowdhry, B. Z. eds.), Oxford University Press, pp 287-318 (2000).
Freire, E. Differential Scanning Calorimetry. Methods Mol. Biol. 40, 191-218 (1995).
Privalov, P.L. and Potekhin, S.A. Scanning microcalorimetry in studying temperature-induced changes in proteins. Methods Enzymol. 131, 4-51 (1986).
Plum, G.E. and Breslauer, K.J. Calorimetry of proteins and nucleic acids. Curr. Opin. Struct. Biol. 5, 682-690 (1995).
Clas, S.D. et al. Differential scanning calorimetry : applications in drug development. Pharm.Sci.Technol.Today 8, 311-320 (1999).
Clausse, D. et al. Morphology characterization of emulsions by differential scanning calorimetry. Adv. Colloid Interface Sci. 117, 59-74 (2005).
Beezer, A.E. et al. Pharmaceutical microcalorimetry: applications to long-term stability studies. Int. J. Pharm. 179, 159-165 (1999).
Jelesarov, I. and Bosshard, H.R. Isothermal titration calorimetry and differential scanning calorimetry as complementary tools to investigate the energetics of biomolecular recognition. J. Mol. Recognit. 12, 3-18 (1999).
Plotnikov, V.V. et al. A new ultrasensitive scanning calorimeter. Anal.Biochem.250, 237-244 (1997).