请了解如何在药品发现过程中使用等温滴定量热法 (ITC) 来调节蛋白质间相互作用
蛋白质间相互作用为很大一类药品开发的定义了有效靶标,其中包括癌症、炎症、糖尿病、骨质疏松症、感染和自身免疫性疾病。截至目前,蛋白质间相互作用受到了破坏或被其他蛋白质所取代。例如,抗体或嵌合构建体被用来抑制配体/受体相互作用,而重组蛋白质被用来替代有缺陷或不足的天然蛋白质。使用小分子作为蛋白质间相互作用的拮抗剂或激动剂,这一做法目前仍处于早期阶段。蛋白质间相互作用的成功定位需要对蛋白质伴侣、其相互作用以及该相互作用的变构作用进行精确描述。正如本章节所述,微量热法非常适合运用于进行此种表征。
自从 Privalov 在 70 年代作出的开创性工作以来,1-4 差示扫描量热法 (DSC) 一直用于确定稳定蛋白质天然结构的作用力大小。经过适当的数据处理,DSC所测得的数据表示的是蛋白质的热容量与温度的关系。下图 1 显示了典型的单体球状蛋白质的扫描结果,它经历了一个两态转变(更复杂的转变,如多态或不可逆变性也可以被准确地处理)5-8。蛋白质中相互作用的破坏引起了一个变性吸热峰,其最大值围绕转变温度 (Tm),这通常被用作衡量蛋白质稳定性的指标。该图显示了在变性完成时,基线向更高热容的典型 S 函数偏移。与该过程相关的焓 (ΔH) 对应于减去 S 型基线后的曲线下面积。此种不利的转变焓与大的有利熵变化 (ΔS) 相对立,后者与受限氨基酸展开时获得的自由度增加有关。这种构象熵的增加足够大,也足以克服在展开时与蛋白质核心水化有关的不利的溶解熵。
此外,当疏水残基暴露时,水分子会形成具有高热容的高度结构化水合层。变性的 S 型基线变化对应于这种热容的较大且正向的变化 (ΔCp)。由于热容变化的数值较大,蛋白质变性的焓和熵的变化都对温度有很强的依赖性。图 1 中的插图显示了焓和温度之间的关系。
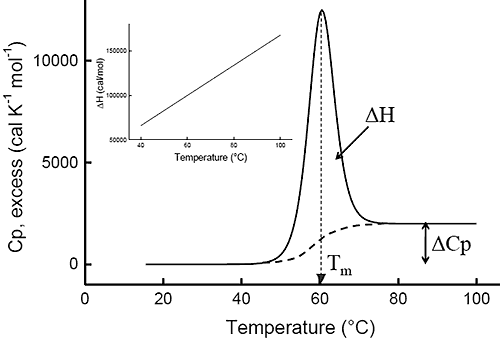
|
蛋白质天然状态的稳定性是由其吉布斯能 (ΔG) 的大小所决定,吉布斯能由标准热力学表达式给出:
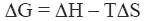
|
其中 ΔH 和 ΔS 分别为焓变和熵变。在本章中,我们将遵循使用天然状态作为参考状态的常见做法。由于焓变和熵变均不是常数,而是温度的递增函数,所以蛋白质稳定的吉布斯能需要写成:
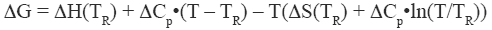
|
其中 TR 是一个方便的参考温度,ΔCp 是热容变化,ΔH(TR) 和 ΔS(TR) 是该温度下的焓和熵的数值。因为 60°C (333.15 K) 接近蛋白质的中位变性温度,其被选为方便的参考温度以比较几种蛋白质,因为该参考温度可使外推误差最小。
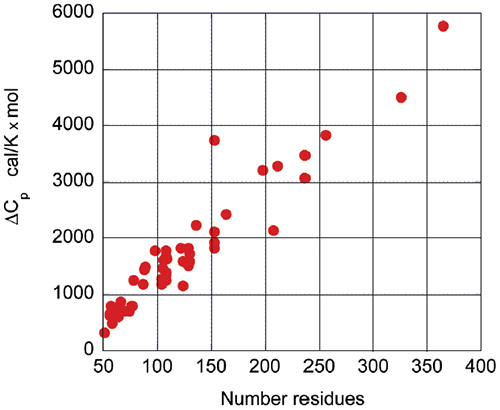
与球状蛋白质的折叠/展开相关的能量学表现出很大的规律性,反映了稳定天然结构的作用力的共同性质。比较不同球状蛋白质在 60°C 的焓变时,可以看到焓变与蛋白质的大小(残基数)之间存在明显相关性。图 2 说明了这一现象,为解释新型蛋白质的热力学数据提供了重要基准。例如,比给定大小的预期焓变更小的焓变表明蛋白质可能存在非结构化区域。
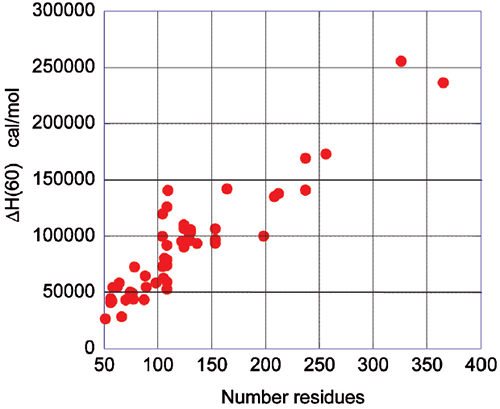
|
在蛋白质展开时,焓变的主要影响因素来自于分子内相互作用(范德华力、氢键等)的破坏以及平行发生的相互作用基团的溶剂化。平均而言,这些影响因素对于不同的球状蛋白质来说是相似的,因此以大小为标准。次要影响因素(如与质子化/反质子化过程有关的影响因素)不足以解释焓变或熵变大小的巨大偏差。如图 3 所示,展开时的热容变化也与蛋白质大小相关。
|
热容变化主要是由于基团水化的改变,天然状态下这些基团被隐埋在溶剂中,而在展开时变成暴露于溶剂中。多年来中所周知的是,与水化有关的热容变化是局部性的,并以溶剂可接触的表面积的变化为尺度 9-12。对于球状蛋白质而言,天然状态下被隐埋的表面积与蛋白质中残基的数量成正比。因此,焓变和热容变化可用于评估蛋白质的结构水平。
文献中报告的最值得注意的案例之一对应于 HIV-1 包膜糖蛋白质 gp120(参考文献 13 和本实验室未发表的工作)。
gp120 是一个由大约 500 个氨基酸组成的蛋白质,它经历两态展开,Tm 为 59.2°C,这个值接近普通球状蛋白质的预期值。与过渡有关的焓变是 180 kcal/mol,转化后的数值仅为 2.3 cal/g。这还不到一般球状蛋白质在相同温度下预期值的一半,相当于大约 300 个残基蛋白质的展开。Tm 值和协同两态转变表明,gp120 存在一个结构化良好的核心和结构化非常低的区域,这些区域对展开过程的焓值没有作用。
在对热力学参数进行分子解释之前,有必要评估所研究的蛋白质是否具有分析所需的质量。在这里,DSC 也可以发挥关键作用。表征良好的蛋白质批次可以作为评估未来批次质量的参考。高质量的重组蛋白质不仅需要不含杂质,还需要不含同一蛋白质的错误折叠、变性或部分变性的形式。传统分析技术(如凝胶电泳、光散射或质谱分析),无法检测或定量分析溶液中错误折叠的蛋白质形式。对于酶而言,特异性比活度测量或活性位点滴定可以对非活性形式的酶进行评估,但对于缺乏酶活性的蛋白质,这是一项非常困难的任务。通常,需要同时采用多种技术对蛋白质进行表征,包括色谱、电泳、质谱、核磁共振 (NMR)、DSC 等。一旦获得高质量的、表征良好的蛋白质,可以将其作为标准品。DSC 提供了一种有效和快速的方法,通过与标准品进行比较来准确评估一批蛋白质。
一批表征良好的蛋白质(标准品)的 DSC 通过三个不同参数加以表征:1) Tm 或最大峰的位置;2) 焓或曲线下面积;3) 曲线形状。对于想要达到相同质量的新的一批蛋白质,应当在这三个参数上符合标准。Tm 对配体或辅因子的存在较为敏感,因此,Tm 的上下变动可能表明存在或不存在配体或辅因子。曲线下面积是衡量蛋白质是否存在变性形式的最佳指标。如果面积较小,则可以清楚表明并非所有蛋白质都正确折叠。事实上,正确折叠蛋白质的比例可以通过新蛋白质和标准品的测定面积比值进行估算。曲线形状反映了折叠/展开机制,因此,如果存在部分折叠形式,也会发生热变性,从而影响曲线形状。图 4 显示了不同批次 gp120 获得的 DSC 结果的示例。每个批次的曲线下面积与正确折叠 gp120 的比例成正比,并等于通过 ITC 确定的能够结合 CD4 的比例。

|
蛋白质伴侣经单独表征后即可评估和解释其相互作用的性质和影响。需要考虑不同的情形,所有这些情形都具有不同的热力学特征。例如,一种可能的情形是两个结构化良好的蛋白质结合而不引起任何重大的构象变化;第二种可能的情形是两个结构化良好的蛋白质结合并引起构象变化;第三种可能的情形是一个蛋白质与另一个蛋白质结合,其非结构化区域由于结合而变得结构化。所有这些情形都可以结合 DSC 和 ITC 数据进行解释;或者在更复杂的情形下,纳入结构数据和结构参数化热力学来进行解释。14-16
在没有构象变化的情况下,两个蛋白质的结合能量由其发生相互作用的表面所主导。焓变大小和符号取决于两个表面之间相互作用的性质。不同蛋白质的文献记录值从放热焓到吸热焓不等。由于结合时被溶剂隐埋区域主要为疏水,两个蛋白质结合与热容的负向改变相关。疏水表面的隐埋也与正向熵变相关,在没有构象结构化的情况下,会产生有利的净熵变。
如果结合引起明显的蛋白质结构化,情况会非常不同。在这种情况下,结合热力学不受发生相互作用的表面所主导,而通过 X 射线晶体学可以观察发生相互作用的表面。我们将再次用 HIV-1 包膜糖蛋白质 gp120 与其细胞表面受体 CD4 的结合来对此进行说明。这是 HIV-1 感染的第一个事件,这种相互作用是药物开发的一个主要靶标。如上所述,gp120 在非配体状态下是一个基本非结构化的蛋白质,然而晶体结构仅存在于 gp120 与 CD4 复合物以及 17b 或 X5 任一单克隆抗体的 Fab 片段中,其中 N 端和 C 端被截断,大部分可变环被去除。17,18
Myzska 等人 19 进行的首次 ITC 研究表明,CD4 受体的可溶性形式与 gp120 的结合与异常大的有利焓变相关,而这种焓变与大的不利熵变相反。该实验室后续研究提供了结合反应的完整描述 13。在此情况下,基本上非结构化的 gp120 分子与 CD4 结合时发生了大规模结构化,引起了异常大的负向热容变化,反映出大量氨基酸被水隐埋。
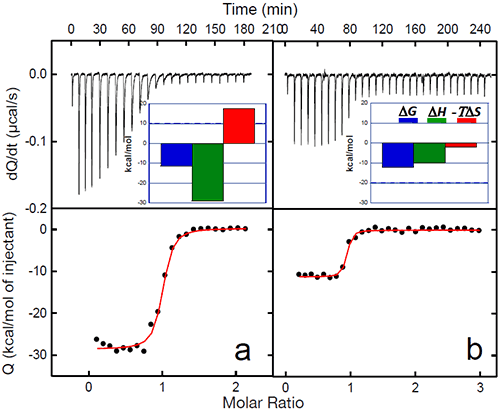
一种蛋白质与另一种蛋白质的结合往往会引发构象变化,从而启动信号级联放大。就 gp120 而言,与 CD4 的结合会引起结构化,从而触发 gp120 中趋化因子协同受体部位的组织和相应功能。CD4 对结合位点的组织消除了对协同受体(CCR5 或 CXCR4)的能量损耗,产生更高的结合亲和力。CD4 与 gp120 的结合引发了与协同受体的结合以及随后导致病毒和细胞膜融合的一系列事件。该实验室通过 ITC 使用单克隆抗体 17b 对 CD4 诱导的协同受体位点的亲和力增加进行了深入研究,该单克隆抗体可以与趋化因子受体 CCR5 的结合位点重叠的表位结合 13,20。MAb 17b 以 5 nM 的亲和力与 gp120 结合,该过程的特点是较大的有利焓变 (ΔH= -28.9 kcal/mol) 以及相反的较大的不利熵变 (ΔS= -59 cal/(K• l mol))。CD4 与 gp120 结合会增加 17b 的亲和力,后者以亚纳摩尔的亲和力与 gp120-CD4 复合物结合。亲和力增加与较小的焓变 (ΔH= -10.1 kcal/mol) 和略微有利的熵变 (ΔS= 6.7 cal/(K • mol)) 相关。较小的焓变反映没有出现大的结构化(其已经由 CD4 的事先结合完成),有利的熵变则反映去溶剂化不再与构象自由度的较大损失相关。下图 5 显示了 17b 单独与 gp120 的结合以及与 CD4 的结合。对协同受体亲和力的变化(由 17b 模拟)可以通过 CD4 与 gp120 结合的热力学来定量解释。CD4 与 gp120 结合时,焓变和热容变化分别为 -34.6 kcal/mol 和 -1.8 kcal/(K • mol);其数值大小让人联想到蛋白质折叠而非结合反应(图 2 和图 3)。事实上,通过使用热容变化的数值结合不利的熵变 -79 cal/(K • mol),可以估计 gp120 中接近 130 个残基在与 CD4 结合时变为结构化(参见参考文献 14)。
|
gp120 中的协同受体位点在未配位蛋白质中基本上为非结构化,17b 的直接结合受到所需结构的阻碍。gp120 中的协同受体位点在与 CD4 结合后变为结构化并具有结合能力,从而消除了对协同受体的结构化要求,因此,结合亲和力明显更高。
竞争性抑制剂能够与配体蛋白质的相同结合位点进行结合,阻止其结合。在开发这类抑制剂时,存在优点和不足。其主要优点在于结合位点结构通常已知,有利于开展基于结构的设计。但存在两点主要不足:1) 对极高亲和力的要求;2) 抑制剂作为替代配体的可能性。由于蛋白质间相互作用常常表现出高亲和力,破坏这类相互作用需要非常高亲和力的抑制剂,而小分子难以实现此种效果。此外,由于与原始配体结合位点相同,抑制剂最终诱发与蛋白质配体相同效果的可能性始终存在。对于 CD4/gp120 相互作用的一些竞争性抑制剂,已经报告存在较为明显的此种情况。
NBD-556 是最近发现的一种低分子量化合物 21,其可以与 gp120 结合并阻断与 CD4 的相互作用 20。NBD-556 与 gp120 结合的亲和力为 3.7 μM,即大约比 CD4 低三个数量级。尽管 NBD-556 与 gp120 的亲和力很低,但其与 gp120 的直接结合却伴随着较大的有利焓变和不利熵变以及较大的负向热容变化,这种热力学特征与 CD4 结合时观察到的相似。在实验中研究了 NBD-556 与 CD4 竞争的能力,存在和不存在 NBD-556 的情况下,对 gp120 与 CD4 结合进行滴定。存在竞争性抑制剂的情况下,采用以下方程可以较为容易地计算预期表观亲和力:

|
其中 Ka 是不存在抑制剂时 sCD4 的结合常数,Ka, app 是存在抑制剂时 sCD4 的预期表观结合常数,Ka,I 是抑制剂的结合常数,[I] 是抑制剂的浓度。采用以下公式计算存在抑制剂时的预期焓变 ΔHapp:

|
其中 Fb 是 gp120 与预结合的抑制剂的饱和程度,ΔH 和 ΔHI 分别是与 sCD4 结合以及与抑制剂结合的焓变。
存在 NBD-556 的情况下,从 sCD4 的 ITC 实验中获得的实验值可以很好地用方程 3 和 4 来解释,这表明 NBD-556 和 CD4 竞争结合 gp120 中的相同位点。与 CD4 的情况一样,NBD-556 与 gp120 的结合使协同受体位点结构化,增强 MAb 17b 的结合亲和力,其在细胞分析中的作用类似于 CD4 的替代物。事实上,在 CD4 阴性细胞中,NBD-556 的存在使 HIV-1 的感染率提高了三个数量级 20。这一结果清楚地说明了竞争性抑制剂的危险性,它在所有方面都能模拟天然配体。
变构抑制剂不会与蛋白质配体竞争;相反,它们通过与变构途径上的某个位点结合来抑制信号级联放大的传播。与竞争性抑制剂一样,变构抑制剂也存在优点和不足。主要优点是其不必取代高亲和力的蛋白质配体,因此对其亲和力要求不严格。主要不足在于,很多时候变构作用的位点未知或者不是立即可见。
BMS-378806 (BMS-806) 及其类似物 22-24 是低分子量 gp120 拮抗剂的示例,通过一种变构机制来抑制 HIV-1 感染。
BMS-806 对 HIV-1 感染具有强效和特异性抑制作用,其对大多数 HIV-1 毒株的 IC50 小于 10 nM。
由于 BMS-806 在高通量感染性筛选时被选出,其确切的进入抑制机制一直存在争议。最初认为 BMS-806 主要通过阻断 gp120-CD4 的相互作用而发挥作用 23,24。然而,开展的量热研究和病毒抑制试验一致表明其具有不同的作用方式 20,25。BMS-806 对 gp120 的结合亲和力为 43 nM,该过程主要是熵驱动 (ΔS= 30 cal/(K • mol)),同时焓变接近零。这些数值对于小分子量抑制剂的结合而言并不罕见。BMS-806 与 gp120 结合时的热容变化为 -292 cal/(K • mol),这是一个小疏水分子被隐埋的典型值。在不存在和存在 BMS-806 的情况下,gp120 与 CD4 结合滴定的 ITC 实验结果与假设存在竞争性抑制的预期值具有较大偏差(方程 3 和 4)。CD4 与 BMS-806 饱和的 gp120 的亲和力仅降低 10 倍,而根据方程 3,如果 BMS-806 和 CD4 竞争 gp120 中的同一位点,则亲和力预期会降低 7000 倍。ITC 结果与病毒抑制试验一致,后者证实 BMS-806 也能抑制 CD4 阴性细胞的 HIV-1 感染。
HIV-1 糖蛋白质 gp120 的例子和开发 HIV-1 进入抑制剂的努力清楚地说明了与开发小分子量蛋白质间相互作用抑制剂有关的问题。
一个能竞争性地阻断 CD4-gp120 相互作用的抑制剂需要具有极高亲和力,从而能够取代亲和力约为 10 nM 的 CD4。此外,CD4 的竞争性置换还要求配体结合时不触发最终导致增强协同受体结合和病毒进入的构象变化。NBD-556 能竞争性抑制 CD4 与 gp120 的结合,但由于其亲和力低,只有在非常高的浓度下才能观察到其对 CD4 结合的影响。NBD-556 的作用也类似于 CD4 模拟物,能够引发 gp120 发生相同的构象变化,导致协同受体激活,从而产生非常严重的不利作用。
BMS-806 对 gp120 的亲和力略低于 CD4,然而 BMS-806 是一种变构抑制剂,不需要与 CD4 竞争就能实现对病毒进入的有效抑制。
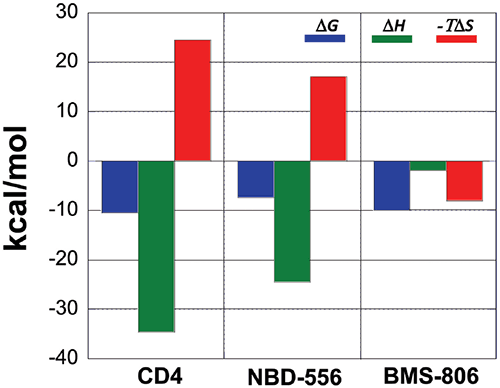
不同抑制剂的结合热力学的比较可以作为选择和优化蛋白质间相互作用的抑制剂的指导。图 6 显示了 CD4、NBD-556 和 BMS-806 与 gp120 结合时的热力学特征。由于 CD4 与 gp120 的结合与 gp120 的大量结构化相关,其热力学特征非常明显,由较大的负向焓变构成,与较大的不利熵变相反。任何与 CD4 竞争并引发类似构象变化的抑制剂也会激活协同受体位点,并表现出与 CD4 观察到的类似的热力学特征。NBD-556 即为此类抑制剂的示例。
不会诱导 gp120 结构化的抑制剂将不会表现出这种热力学特征。例如,与不同位点结合而不诱发任何构象变化的变构抑制剂 BMS-806,由于其完全不同的热力学特征,很容易加以区分。此外,一种与 CD4 竞争而不引起 gp120 结构化的抑制剂将具有接近 BMS-806 的热力学特征,即以较小甚至有利的熵变为特征的热力学特征。在此方面,我们已经能够确定,可用于提高结合效力的位点不一定与启动变构效应的位点重叠。
使用诸如 CORE_BIND 26 或类似算法来区分这些位点,应当能够开发出不会作为替代配体的竞争性抑制剂。
|
本研究工作获得美国国立卫生研究院(GM56550 和 GM57144)和美国国家科学基金会 (MCB0641252) 的资助。